US Pharm.
2008;33(1):HS-23-HS-30.
Antiphospholipid syndrome (APS) is an autoimmune disease characterized by the
presence of antiphospholipid antibodies, manifesting with vascular thrombosis
and/or recurrent fetal loss.1 It is a complex illness that
paradoxically causes either too much coagulation or too little, although
thrombosis is most commonly seen.
Antiphospholipid antibodies (aPL) are a heterogeneous group of autoantibodies
directed against phospholipids, a group of molecules that make up most cell
membranes. Phospholipids are bound to, and circulate with, positively charged
phospholipid-binding proteins. Cell membranes are involved in many biological
functions, including coagulation. Several key reactions involved in blood
clotting require membranes that contain certain negatively charged
phospholipids.
Primary APS occurs in patients without clinical evidence of another autoimmune
disease, whereas secondary APS occurs in association with connective tissue
disorders, most commonly systemic lupus erythematosus (SLE), or another
rheumatic or autoimmune disorder. About 50% of patients have the primary form
of the disease.2 Primary APS occurs more commonly in young- to
middle-aged adults; however, it also manifests in children and elderly people.
Disease onset has been reported in children as young as 8 months. A female
predominance is documented, particularly for secondary APS. Secondary APS is
present in 10% to 15% of patients with SLE.3,4
Antiphospholipid Antibodies Associated with APS
The aPL are a heterogeneous group of autoantibodies that may be present in
healthy persons. Prevalence varies between 0% and 9% depending on the aPL, but
positive findings seldom persist.2 These antibodies rise
transiently in acute settings and are especially prevalent in thrombosis. The
antigenic targets of aPL are uncertain. Although the name implies that
phospholipids such as cardiolipin are the targets, the major antigens are most
likely phospholipid-binding proteins, or protein cofactors, bound to
phospholipids. The most important protein cofactor in APS is ?2
-glycoprotein I (?2GPI; also called apolipoprotein H). ?2
GPI appears to be an in vivo anticoagulant, interfering with the contact
activation of the intrinsic pathway in the coagulation cascade.1 ?
2GPI is bound to cardiolipin (CL) and lupus anticoagulant (LA), among
other phospholipids. Other phospholipid-binding proteins include prothrombin,
protein C, protein S, factor VIII, and annexin V. Many of these protein
cofactors play a role in regulating coagulation. Binding by aPL interferes
with normal function and may lead to a procoagulant state.
The aPL may also be present in patients with infections such as syphilis,
infectious mononucleosis, AIDS, and exposure to certain medications (e.g.,
chlorpromazine). These aPL are not believed to have clinical sequelae because
they bind directly to phospholipids rather than to phospholipid-binding
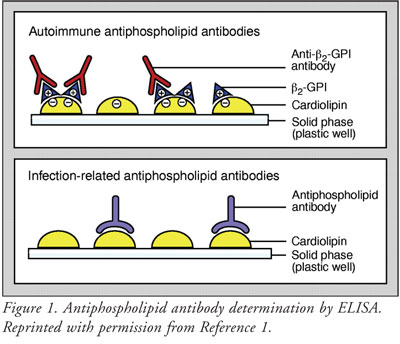
proteins. The aPL can be detected by enzyme-linked immunosorbent assay
(ELISA), using plastic wells coated with negatively charged phospholipids.
ELISA, however, may fail to distinguish aPL that bind to phospholipids as
opposed to phospholipid-binding proteins (FIGURE 1).1
The most important subgroups of aPL
associated with APS are LA, anticardiolipin antibodies (aCL), and anti-?2
GPI antibodies. Detection of these aPL are complicated by poor standardization
of tests.
LA is an antibody specific to either phospholipid-bound prothrombin or ?2
GPI.5 In patients with SLE, LA correlates particularly well with
thrombosis. LA blocks phospholipid surfaces integral to coagulation.2
By inhibiting the conversion of prothrombin to thrombin, LA interferes with
clot formation. This interference is measurable by coagulation assay,
activated partial thromboplastin time (aPTT), presenting as prolonged clotting
time.6 In order to confirm the presence of LA, international
consensus criteria recommend performing at least two different types of assay
for LA, with concurring results. The most common LA tests are aPTT and the
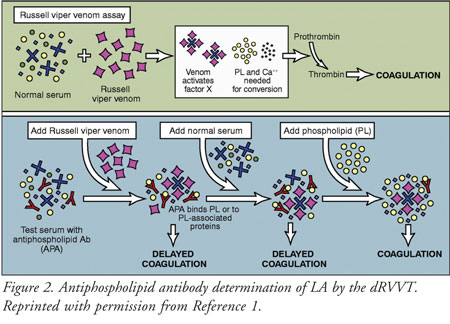
dilute Russell viper venom time (dRVVT). The dRVVT is performed by adding
Russell viper venom to serum. Normally, the venom activates the coagulation
cascade and coagulation occurs. A positive result occurs when: 1) coagulation
is prolonged instead; 2) adding normal serum fails to correct this; but 3)
normal coagulation is resumed by the addition of phospholipids (FIGURE 2
).7 Anticoagulant therapy interferes with the detection of LA, and
testing should be postponed if treatment is under way.8
The aCL bind to cardiolipin, a phospholipid, through ?2GPI, and are
detected by ELISA.5 Notably, aCL that occur with syphilis bind
directly to cardiolipin in the absence of ?2GPI. This binding is
inhibited by human ?2GPI and its antibody (FIGURE 1) and
highlights the difference between aCL associated with thrombosis and aCL
associated with infection. The immunoglobulin isotypes associated with aCL are
IgG, IgM, and IgA. It is believed that the IgG isotype is most strongly
associated with thrombosis.2 Current criteria for the
classification of APS recommend the use of ELISA to measure ?2
GPI-dependent anticardiolipin IgG and IgM antibodies (TABLE 1).7

Anti-?2GPI antibodies were added to the modified Sapporo criteria
for APS diagnosis. As noted earlier, ?2GPI bound to negatively
charged phospholipids is the major target for aPL. Anti-?2GPI
antibodies are an independent risk factor for thrombosis and obstetric
complications.6,9 Current criteria for classifying APS also
recommend ELISA to measure anti-?2GPI IgG and IgM antibodies (
TABLE 1).
Pathogenesis
The presence of aPL alone does not
necessarily have clinical implications, but in some people, aPL may trigger
thrombosis. One mechanism for aPL-associated thrombosis involves binding of ?
2GPI by autoantibodies, which in turn facilitates binding to membrane
phospholipids and/or phospholipid-associated receptors. The resulting anti-?
2GPI/?2GPI complex activates platelets, monocytes, and
endothelial cells through binding of ?2GPI with specific cell
surface receptors (FIGURE 3), leading to a procoagulant state. Other
proteins that are important in regulating coagulation (i.e., prothrombin,
protein C, protein S, and annexin V), may also be targeted by aPL.6
Another mechanism involves the binding of aPL to endothelial cells, which
induces activation and promotes expression of cytokines and metabolism of
prostacyclins.6 Activation of endothelial cells promotes
coagulation.10 There is also evidence that aPL promote the
activation and aggregation of platelets. Finally, thrombosis in APS has been
linked to heparin-induced thrombocytopenia.6 In APS, there is a
high rate of recurrence of similar thrombotic events. A "second hit"--such as
an infection leading to cytokine production or traumatic injury to the
vascular bed leading to endothelial activation--and nonimmunologic
procoagulant factors are necessary for thrombosis to occur.1
Diagnostic Criteria
An international consensus statement on the preliminary classification
criteria for APS was published in 1999, called the Sapporo criteria.11
These criteria were updated in 2006.7 A diagnosis of APS is made
when a person presents with at least one of the clinical and at least oneÜ of
the laboratory criteria. Specifically, these are: 1) the occurrence of
clinical manifestations, such as vascular thrombosis or obstetric
complication; and 2) the persistent presence of aPL. The aPL specified in the
consensus criteria are LA, aCL (medium-to-high titer IgG and/or IgM isotype),
and ?2GPI antibodies (high titer IgG and/or IgM isotype). To
demonstrate that the results are persistent, positive laboratory results must
be confirmed on two or more occasions, at least 12 weeks apart. The qualifying
clinical criteria and quantitative laboratory findings are summarized in
TABLE 1.7
Management of APS
Prophylaxis in aPL-positive Patients:
Given that laboratory
criteria for APS are not routinely performed, APS is usually identified during
the differential diagnosis of one of the clinical criteria. Hence, treatment
is generally considered in aPL-positive patients after a thrombotic event
occurs. If aPL are identified in asymptomatic patients, any factor
predisposing for thrombosis (e.g., defects in coagulation factors, platelet
defects, hyperviscosity, oral contraceptives, estrogen replacement therapy,
nephrotic syndrome, smoking, or surgery) should be corrected, if possible.
The role of aspirin for primary prophylaxis against a thrombotic event is
controversial. One report found that aspirin (325 mg/day) in women who are aPL
positive and with a history of fetal loss may protect against thrombosis.
12 On the contrary, a case-control study within the Physicians
Health Study found that aspirin (325
mg/day) did not protect against venous thromboembol
ism in males with aCL.13 Currently, there are insufficient data to
recommend antithrombotic prophylaxis in patients who are aPL positive without
a history of thrombosis. In women who are aPL positive and with a history of
fetal loss, however, prophylaxis with aspirin may be considered.
Clinical Manifestations and Management
Vascular Thrombosis:
The defined clinical
criteria are arterial, venous, or small vessel thrombosis, excluding
superficial thrombi.7 Venous thromboembolism is the most common
initial clinical manifestation among patients with APS, occurring in 32% who
meet Sapporo criteria.2 The most common form is deep venous
thrombosis of the lower extremities. Up to half of those patients have
pulmonary emboli.6 Arterial thromboses are most common within the
cerebral vasculature, and features are consistent with ischemia and
infarction.
Other manifestations that are considered clinical features associated with
APS, but are not diagnostic, include cardiac valve abnormalities, myocardial
infarction, coronary artery disease, pulmonary hypertension, thrombocytopenia,
hemolytic anemia, nephropathy, neurologic manifestations, and livedo
reticularis, a lattice-like pattern of superficial veins most often found on
the thighs, shins, and hands. Presence of one or more of these features,
especially with laboratory criteria, raises suspicion of APS.7
Initial treatment of venous thrombosis is the same, regardless if APS is
implicated. Laboratory tests to rule out APS should be performed prior to
initiation of anticoagulation therapy, and other thrombophilic causes
excluded. Intravenous heparin or low-molecularñweight heparin (LMWH) for at
least five days should be initiated, overlapped with warfarin therapy until a
target international normalized ratio (INR) of 2.0 to 3.0 is achieved and
maintained.1,14,15
For long-term treatment, low-dose aspirin alone or low-intensity warfarin is
ineffective in preventing recurrence. In a retrospective study among 70
patients with APS, warfarin treatment of intermediate intensity (INR 2.0 to
2.9) and high intensity (INR 3.0 or higher) significantly reduced the rate of
recurrent thrombosis, whereas low-intensity treatment (INR 1.9 or less) did
not confer significant protection.16 In this trial, there were five
nonfatal bleeding complications. The evaluators did not report which event
happened during which treatment.16
To further clarify if APS should be treated with intermediate or
high-intensity warfarin, a randomized, double-blind clinical trial was
performed in 114 patients with aPL and previous thrombosis.
Intermediate-intensity warfarin (INR 2.0 to 3.0) was superior to
high-intensity warfarin (INR 3.1 to 4.0) for thromboprophylaxis. The incidence
of recurrent thrombosis was 10.7% in patients assigned to receive
high-intensity warfarin and 3.4% in patients assigned to receive
intermediate-intensity warfarin. Eleven patients (19%) in the
intermediate-intensity group and 14 patients (25%) in the high-intensity group
had at least one episode of bleeding, although the difference was not
statistically significant.17
The efficacy findings suggest that intermediate-intensity warfarin is
appropriate for patients with APS. Although differences in bleeding rates were
not significant in this trial, higher INR targets generally increased risk for
hemorrhagic complications.18
In persons with recurrent
thromboses despite warfarin treatment, adjusting the warfarin dosage to target
a higher INR (3.0 to 4.0) and adding low-dose aspirin are recommended.14
Normalization of the LA or aCL is not an indication to discontinue
anticoagulation, because patients remain at risk for new thromboses regardless
of change in titer. There is a higher risk of recurrent thromboses in the six
months following discontinuation of warfarin.4 Thus, APS patients
with vascular events should remain on warfarin therapy indefinitely.15
Arterial thromboembolism in APS affects the cerebral circulation, manifesting
as stroke and transient ischemic attacks, and with less certainty may manifest
as myocardial infarction. Treatment for patients with APS and a first ischemic
stroke consists of anticoagulation with aspirin (325 mg/day) or
intermediate-intensity warfarin (INR 1.4 to 2.8). This was based on a
prospective cohort study performed in 1,770 patients with presence or absence
of aPL, comparing intermediate-intensity warfarin and aspirin (325 mg/day) for
prevention of recurrent stroke or death. The investigators found no difference
in the risk of thrombotic events in patients treated with warfarin compared
with aspirin, as well as no difference in the risk of bleeding.19
Treatment of APS with recurrent thrombotic events despite warfarin therapy is
uncertain. These persons may be treated by increasing the target INR (2.5 to
3.5 or 3.0 to 4.0), switching from warfarin to therapeutic dosages of
unfractionated heparin or LMWH, or adding an antiplatelet agent to warfarin.
2
Obstetric Complications
Pregnancy losses in APS typically occur at or after the 10th week of
gestation. It is hypothesized that APS-related pregnancy loss results from
poor placental perfusion due to localized thrombi.6 In addition,
the reduction of annexin V in the placenta (previously known as placental
anticoagulant protein I) may also be an important mechanism of thrombosis and
pregnancy loss in APS.20 The aPL reduce levels of annexin V and
accelerate the coagulation of plasma. Elevated estrogen levels during
pregnancy are also associated with increased risk of thrombosis, even in the
absence of aPL.21
Women with aPL and repeated pregnancy loss, but no history of thrombosis or
SLE, can achieve a similar live birth rate as that of non-aPL positive women
(approximately 80%) with the use of either low-dose aspirin alone or heparin
(5,000 units every 12 hours) or LMWH (enoxaparin, 1 mg/kg or 40 to 80 mg;
dalteparin 5,000 units, administered once daily) plus low-dose aspirin.
2,6,22
In women with previous
thrombosis and fetal loss, those who fulfill the Sapporo criteria should
receive low-dose aspirin (81 mg), full anticoagulation with subcutaneous
unfractionated heparin (10,000 units every 12 hours), or LMWH.14
Treatment should begin as soon as intrauterine pregnancy is documented. LMWH
should be discontinued at the 36th week of gestation and replaced by
unfractionated heparin. Thromboprophylaxis should be continued for six to
eight weeks after delivery.23
Women who desire to become pregnant and are taking warfarin prior to pregnancy
should be counseled to switch to heparin either before conception or as soon
as pregnancy is confirmed to avoid the risk of warfarin embryopathy.23
Patients with high-titer aPL have about a 50% to 75% chance of fetal loss, but
with aspirin and heparin the chance of full-term delivery increases 70% to 80%.
24
Intravenous immunoglobulin (IVIg) is an immune globulin currently used for the
treatment of immune thrombocytopenic purpura, Guillain-BarrÈ syndrome,
Kawasaki disease, and polymyositis/dermatomyositis. Its role in APS is to
inhibit aPL. This may be due to the presence of anti-idiotypes to aPL within
IVIg preparations or to the presence of F(ab)2 fragments from IVIg
that inhibit the binding of aCL to cardiolipin in a dose-dependent manner.
IVIg infusions at varied dosages (400 mg/kg/day for five days; alternatively,
1 to 2 g/kg in divided doses over two to five days given monthly), at varied
time of administration, and with concomitant therapies (e.g., heparin,
aspirin), have been reported in patients with aPL who continue to lose
pregnancies despite receiving low-dose aspirin and heparin.1,25
Reports have suggested that the mechanism of action of IVIg in the treatment
of APS involves short-term neutralization of aPL, resulting in a long-term
decrease in antibody titers.25
Caution should be observed in patients with IgA deficiency; serum IgA levels
should be checked prior to IVIg to prevent severe reactions. Adverse effects
may include migraine attacks, 10% increased risk of aseptic meningitis, and
increased risk of urticaria, pruritus, or petechiae two to five days after
infusion.25
Catastrophic APS
A minority of patients may present with a life-threatening syndrome called
catastrophic APS (CAPS). It is diagnosed when evidence of involvement of three
or more organs within one week is confirmed by histology evidence and the
presence of aPL. Venous or arterial thrombosis of large vessels is less common
in patients with CAPS.6 The kidney is the organ most commonly
affected, followed by the lungs, the heart, and the skin.6
Disseminated intravascular coagulation may also occur specifically in CAPS,
but not in primary or secondary APS. Infections, surgical procedures,
withdrawal of anticoagulant therapy, and the use of drugs such as oral
contraceptives are considered precipitating factors for CAPS.
Patients with CAPS are usually treated with full anticoagulation with IV
heparin, overlapped with warfarin therapy to achieve an INR of 3.0. Other
agents that may have a role in treatment of CAPS include corticosteroids,
IVIg, and plasmapheresis.26 With limited evidence, long-term oral
anticoagulation is recommended to prevent further APS-related thrombotic
events.26
Conclusion
APS is an
autoimmune disorder that presents unique challenges in diagnosis and
treatment. The revised diagnosis can be simply stated as the presence of one
clinical and one laboratory criteria. In practice, the heterogeneous
presentation of both clinical and laboratory findings complicates diagnosis.
This disorder should always be included in differential diagnosis of persons
who have coagulation defects, evidence of vascular thrombosis, and/or history
of recurrent miscarriages or fetal losses. Positive laboratory testing should
always be confirmed at least 12 weeks apart to verify persistence.
The heterogeneous
presentation of APS also makes treatment challenging, and, in particular,
research clarifying optimal therapy remains lacking. Currently, the mainstay
of treatment is anticoagulation in those persons who develop an acute
thrombotic event. Although indefinite duration of anticoagulation therapy is
recommended, the decision to administer long-term anticoagulation certainly
requires judicious clinical evaluation and risk assessment, given the
potential hemorrhagic complications of anticoagulation therapy.
References
1. Hanly J.
Antiphospholipid syndrome: an overview. CMAJ. 2003;168:1675-1682.
2. Lim W, Crowther MA, Eikelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA. 2006;295:1050-1057.
3. Hughes G. Thrombosis, abortion, cerebral disease and the lupus anticoagulant. Br Med J. 1983;287:1088-1089.
4. Khamashta M, Cuadrado MJ, Music F, et al. The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med. 1995;332:993-997.
5. Roubey R. Autoantibodies to phospholipid-binding plasma proteins: a new view of lupus anticoagulants and other "antiphospholipid" autoantibodies. Blood. 1994;84:2854-2867.
6. Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med. 2002;346:752-763.
7. Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome. J Thromb Haemost. 2006;4:295-306.
8. Tripodi A, Chantarangkul V,
Clerici M, Mannucci PM. Laboratory diagnosis of lupus anticoagulants for
patients on oral anticoagulant treatment. Thromb Haemost. 2002;88:
583-586.
9. Audrain MA, EI KouriD, Hamidou MA, et al. Value of autoantibodies to b2-glycoprotein 1 in the diagnosis of antiphospholipid syndrome. Rheumatology. 2002;41:550-553.
10. Nawroth PP, Stern DM. Endothelial cell procoagulant properties and the host response. Semin Thromb Hemost. 1987;13:391-397.
11. Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum.1999;42:1309-1311.
12. Erkan D, Lockshin MD. High thrombosis rate after fetal loss in antiphospholipid syndrome: effective prophylaxis with aspirin. Arthritis Rheum. 2001;44:1466-1477.
13. Ginsburg KS, Liang MH, Newcomer L, et al. Anticardiolipin antibodies and the risk for ischemic stroke and venous thrombosis. Ann Intern Med. 1992;117:997-1002.
14. Tektonidou M. Antiphospholipid
syndrome. Orphanet Encyclopedia [serial online]. May 2004. Available at:
www.orpha.net/data/patho/Pro/en/Antiphospholipid-
FRenPro5517.pdf.
Accessed December 18, 2007.
15. Buller HR, Agnelli G, Hull RD, et al. Antithrombotic therapy for venous thromboembolic disease: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126(suppl 3):401S-428S.
16. Rosove MH, Petronella MC, Brewer RN. Antiphospholipid thrombosis: clinical course after the first thrombotic event in 70 patients. Ann Intern Med. 1992;117:303-308.
17. Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med.2003;349:1133-1138.
18. Ansell J, Hirsh J, Poller L, et al. The pharmacology and management of the vitamin K antagonists: the Seventh ACCP Conference on Anthrombotic and Thrombolytic Therapy. Chest. 2004;126(suppl 3):204S-233S.
19. The APASS writing committee. Antiphospholipid antibodies and subsequent thrombo-occlusive events in patients with ischemic stroke. JAMA. 2004;291:576-584.
20. Rand JH, Wu XX, Andree HA, et al. Pregnancy loss in the antiphospholipid-antibody syndrome--a possible thrombogenic mechanism. N Eng J Med. 1997;337:154-160.
21. Erkan D, Yazici Y, Peterson MG, et al. A cross-sectional study of clinical thrombotic risk factors and preventive treatments in antiphospholipid syndrome. Rheumatology. 2002;41:924-929.
22. Bates SM, Greer IA, Hirsh J, Ginsberg JS. Use of antithrombotic agents during pregnancy: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126(suppl 3):627S-644S.
23. Branch DW, Eller AG. Antiphospholipid syndrome and thrombosis. Obstet Gynecol. 2006;49:861-874.
24. Erkan D, Lockshin M.
Antiphospholipid syndrome. Rheumatic Diseases [serial online]. 52(3).
Available at: ww2.arthritis.org/research/Bulletin/Vol52No3/Introduction.
asp. Accessed
December 18, 2007.
25. Sherer Y, Levy Y, Shoenfeld Y. Intravenous immunoglobulin therapy of antiphospholipid syndrome. Rheumatology. 2000;39:421-426.
26. Cervera R, Asherson RA, Font
J. Catastrophic antiphospholipid syndrome. Rheum Dis Clin N Am.
2006;32:575-590.
To comment on this article, contact
editor@uspharmacist.com.



