US Pharm. 2015;40(8)(Pharm&Tech suppl):3-7.
ABSTRACT: The proposed U.S. Pharmacopeia General Chapter 800 (USP <800>) provides standards for protecting personnel and the environment when handling hazardous drugs. While many professional organizations support the USP’s efforts to improve the safety of working environments, critics suggest that the new chapter lacks clarity and leaves room for subjective interpretation. Given the diversity in healthcare settings, these new standards may represent barriers to care for patients served by sites unable to meet these standards. This summary provides key elements of the new chapter for those in practice to review as they form their own opinions.
The United States Pharmacopeial Convention (USP) is a nonprofit scientific organization founded in Washington, DC, in 1820.1 All state societies of medicine were invited to participate in order to create a system of standards and a national drug formulary, which at that time included only 217 drugs. In 1830, the USP’s Committee of Revision published the first revision of the U.S. Pharmacopeia compendium (also called the USP), with planned revisions to continue at 10-year intervals. Current regulations and practice standards are constantly evolving; therefore, the USP compendium is considered a constant “work in progress.”1
U.S. Pharmacopeia
Today, the USP compendium contains thousands of chapters that set standards for the identity, strength, quality, and purity of medicines, food ingredients, and dietary supplements manufactured, distributed, and consumed worldwide. The USP’s drug standards are enforceable in the U.S. by the FDA, and these standards are used in more than 140 countries, providing guidance to scientists and practitioners as they develop and revise standards that help protect public health worldwide. The mission of the USP is to improve global health through public standards and related programs that help ensure the quality, safety, and benefit of medicines and foods.2
Prior to the most recent inclusion of USP General Chapter 800 (USP <800>), there was no existing USP chapter that addressed the compounding of hazardous drugs. USP <800>, originally published in May-June 2013, provides standards for protecting personnel and the environment when handling hazardous drugs (HDs) and will provide overall guidance for the life cycle of handling such drugs. However, due to concerns expressed by stakeholders, USP <800> is scheduled to be republished with clarified wording and will reflect revised guidance documents. Comments were accepted until May 31, 2015. In essence, USP <800> builds upon and complements both Chapters <795> and <797>.3
An organization or healthcare entity, including pharmacies, hospitals, clinics, physician practice facilities, and veterinary offices where HDs are compounded, must develop a comprehensive health and safety program. Such programs must minimally include specific policies outlining the use and availability of appropriate personal protective equipment (PPE), competent staff who demonstrate robust work practices, procedures for waste segregation and disposal, and primary, secondary, and supplemental engineering controls as well as a program to ensure medical surveillance.
Organizational Policies: Identifying Hazardous Drugs
A hazardous drug is defined as a drug with a potential to impair reproduction in humans; cause fetal harm; cause cancer; damage and/or alter genes; or cause toxicity in the organs of humans or animals, even at low doses.3 There is no acceptable level of personal exposure to HDs, so efforts must be made to minimize any potential exposure. It is important to note that the HDs referenced in USP <800> are now divided into three groups, with some overlap: nonantineoplastic drugs, antineoplastic drugs, and drugs with reproductive risks for men or women. This new list is based on the 2014 National Institute for Occupational Safety and Health (NIOSH) List of Antineoplastic and Other Hazardous Drugs in Healthcare Settings.4
Each specific organizational entity must maintain a list of HDs, which should include items on the current NIOSH list in addition to other agents they deem appropriate for inclusion but that are not referenced on the NIOSH list. This list must be reviewed once a year or more frequently when new agents or dosage forms become available. NIOSH also provides criteria used to identify new HDs that become available after the most current NIOSH list has been published. If insufficient information is available to determine the hazardous potential of new drugs, NIOSH recommends treating the drug as hazardous until further information is available to make a more informed determination.4
The 2014 list includes 27 additions to previous versions such as crizotinib, phenytoin, and fluconazole, while removing other drugs, such as tetracycline, that did not meet the criteria for HDs.4 It is important to remember that an assessment of risk is key when it comes to ensuring safety protocols specific to one’s practice site. The facility should make note of the type of HD, dosage form, risk of exposure, packaging, and manipulation for all the HDs it handles.
Types of Exposures
There are a number of possibilities for unintentional exposures to HDs, which include, but are not limited to, ingestion, inhalation, injection, and mucosal absorption (TABLE 1).3 Both clinical and nonclinical staff can experience unintended exposure when touching contaminated surfaces, when cleaning dust or spills, or when involved in the disposal of these substances. When facilities are developing new policies or updating existing policies, all areas of unintentional exposure risks should be considered. Handling HDs at any level of preparation, dispensing, and administration poses risk of this exposure. Access to areas where HDs are stored and prepared must be limited to only authorized staff to avoid unintentional exposure to HDs. Despite best practices, there may still be unintentional exposures to consider and to include in policy.

Personal Protective Equipment
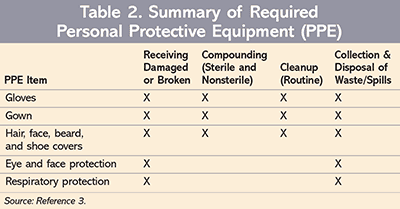
Dosage forms such as capsules and coated tablets may not pose an exposure risk that would be deemed “significant” to healthcare workers unless otherwise altered. However, crushing tablets or preparing solutions of hazardous drugs increases the exposure risk. In order to help healthcare entities managed these exposure risks, the 2014 NIOSH document includes recommendations for the use of PPE in specific situations.4 American Society of Testing and Materials (ASTM)–tested chemotherapy gloves may be required when transporting intact supplies or compounded HDs, or when receiving these products or stocking them.5 However, more intense handling, such as preparing an IV solution of an HD, may require double gloves in addition to eye and respiratory protection, protective gowns, and the use of a biological safety cabinet (BSC) and closed-system drug-transfer device (see TABLE 2 for PPE requirements).3 A single set of gloves may be all the PPE necessary for a healthcare worker to administer an intact capsule or tablet from unit-dose packaging.
Competency and Staff Training: Responsibilities of Personnel Handling HDs
All healthcare personnel who handle HDs are responsible for understanding the fundamental precautions and practices in USP <800>. There is also an expectation that there is an existing process for continual review and reevaluation of policy and procedures to ensure optimal harm prevention for both patients and personnel. This harm prevention must take into account not only immediate personal harm but also harm to the environment. An organizational entity is required to designate a compounding supervisor to be responsible for the development and implementation of these procedures as well as for monitoring and ensuring standards, laws, and regulations.3
Receiving and Storage
Upon receiving HDs, there must be a visual inspection with a plan for subsequent action if damage is suspected to the packaging and/or external shipping container. The unpacking of HDs must be done in a room with negative or neutral pressure with at least 12 air changes per hour (ACPH) in order to reduce spread of contamination in case of leaks and spills. Areas designated for the handling of HDs must be separated from non-HD areas, and the unpacking of HDs from external shipping containers shall not occur in areas used for sterile compounding. HDs received in final manufactured coated-dose forms that are clearly labeled may be stored with non-HDs if the organizational entity includes safety strategies in its standard operating procedures. If safety strategies do not support shared storage, the HDs must be stored separately in a room with negative or neutral pressure with at least 12 ACPH. HDs that require refrigeration must be placed in a designated refrigerator in the HD storage room, buffer room, or containment segregated compounding area (C-SCA). Storage of HDs should be at eye level, not on the floor, and storage containers must limit the risk of leakage and breakage. All HD storage areas and containers must be properly labeled to prevent inappropriate handling.3
Hazard Communication Program
The Occupational Safety and Health Administration (OSHA) requires a written work plan that describes how HDs are identified and defines the process of supplying safety data sheets (SDS) for each HD available within the workplace.3 The entity is also required to address how it will ensure that all employees are trained before any initial exposure or work assignment, and how they will respond to questions or concerns regarding these potential or actual exposures. Robust training and competency assessments must include verification and documentation with specific testing of proper techniques, and these must be reassessed at least annually or more frequently if changes occur or if new products have become available for use. Other considerations for training include, but are not limited to, safety when transporting HDs, compounding different HD dosage forms, and protection when administering HDs. Decontamination, disinfection, disposal, and cleaning as well as actions to take to control spills must also be included in staff training and competency assessments.
Environmental Quality and Control
In order for HDs to be handled appropriately and under conditions that promote safety, there must be administrative, environmental, and engineering controls. Engineering technology is divided into three specific categories: primary, secondary, and supplementary levels of control.3
The International Organization for Standardization (ISO) classes for sterile cleanrooms establish the minimum acceptable threshold to ensure quality, safety, and efficiency. The nature of the planned activities in a specific area defines the ISO cleanroom area classification. For example, USP <797> requires that the area immediately adjacent to the aseptic processing line meet, at a minimum, Class 10,000 (ISO 7) standards under dynamic conditions. Alternatively, an area classified at a Class 100,000 (ISO 8) air cleanliness level is the least restrictive but is still appropriate for the anteroom used for garbing. In critical areas, a Class 100 or ISO 5 is considered to be the highest level of control, appropriate for the area in immediate proximity to exposed sterilized containers/closures and filling/closing operations. Supporting areas, or cleanroom areas where the laminar flow stations are located, must meet the least restrictive Class 100,000 (ISO 8) air quality.3
Containment primary engineering controls (C-PECs) are ventilated devices designed to minimize exposures to HDs through control of airborne contaminants, by providing partial or full enclosure of potential contaminants, using airflow capture to remove airborne contaminants at point of generation, and using air pressure and airflow relationships, as well as high-efficiency particulate air (HEPA) filtration on exhaust streams. Some examples of C-PECs include Class I, II, and III BSCs, compounding aseptic containment isolators (CACIs), and containment ventilated enclosures (CVEs). All C-PECs must be placed in a segregated room with restricted access, with a minimum negative pressure of 0.01 inches of water column, and are required to be externally vented. ISO Class 5 air quality is required for C-PECs used for sterile compounding.3
Containment secondary engineering controls (C-SECs) for the room where the C-PEC is stored must include specific operational parameters necessary to contain potential hazards in the compounding room such as room ventilation and pressurization, access, and barriers. Compounding HDs require C-SEC where any C-PEC shall be vented to the outside through HEPA filtration. A sink and eye wash station for emergency rinsing and washing (from eyes and skin) is required to be readily available for both sterile and nonsterile HD compounding and must meet all applicable regulations. Location of these washing stations is required to be outside of ISO Class 7 buffer areas and must not interfere with required ISO classifications.3
Compounding Technology Requirements
Whether compounding nonsterile or sterile HD products, all C-PECs need to be externally vented and placed in a restricted-access segregated room, which has a minimum negative pressure. As for the C-SEC, the room in general should be vented to the outside air through HEPA filtration. A laminar airflow workbench (LAFW) or CACI should not be used for the compounding of antineoplastic HDs.3
For nonsterile compounding (TABLE 3),3 this section is the same as Chapter <795>, Pharmaceutical Compounding—Nonsterile Preparations. An ISO-classified environment is not needed; therefore, unidirectional airflow is also not required. A Class I BSC, or any other C-PEC or a CVE, can be used for nonsterile HD compounding, but all of them need to be externally vented. If the C-PEC cannot be externally vented, there must minimally be a redundant HEPA filter in the series, though this is not preferred. If dedicated for use with nonsterile compounding, Class II BSCs or a CACI may be used. If a class II BSC or a CACI is usually dedicated to sterile compounding, it can still be used for nonsterile compounding if it undergoes thorough disinfection and cleaning after nonsterile compounding and before reuse for sterile compounding. The C-SEC for nonsterile compounding needs to achieve both minimum ACPH and negative pressure in all adjacent spaces. Where an HD is already a final manufactured product that does not require further manipulation or has no potential for producing aerosolized gasses, a C-PEC will not be required.3

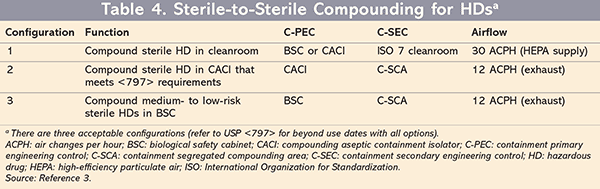
For sterile compounding, there are three configurations for C-PEC/C-SEC combinations (TABLE 4).3 Two of these optional configurations can be used for all types of sterile HDs. For both of these scenarios, the beyond use date (BUD) of the compound will follow the guidelines listed in Chapter <797>. The third option is new and specific to USP <800> for compounding only low- or medium-risk sterile HDs in a BSC. For this configuration, a BSC or a CACI (whether or not it meets USP <797> criteria) will be required in a C-SCA with a minimum ACPH and negative pressure measurements in all adjacent spaces. HEPA filters and ISO 7 are also not required with this low- or medium-risk sterile HD configuration; however, compounds made in this configuration are limited to a BUD of no longer than 12 hours.3
Medical Surveillance
While compounding pharmacies and other facilities are attempting to incorporate the technology requirements of USP <800>, pharmacists must be mindful that a compliant program also takes into account the actual physical health of employees. A medical surveillance program is required in order to routinely monitor the health status of all employees who are potentially exposed to HD. Data collected should be analyzed to evaluate and ensure the protection of good work practices. The surveillance should focus on identifying the earliest signs of reversible damage in order to prevent irreversible outcomes. Any data that show possible adverse effects due to exposure should assist in the identification of failure vulnerability and allow for systematic corrections to prevent future exposures.3
Conclusion
As the healthcare system prepares for the implications of USP <800>, we must consider not only the physical facility requirements, but also the policies, procedures, and training that will soon be enforceable. There should also be an expected ripple effect that will spill over to facilities because of the additional expense and limited options to expand, especially for organizations that may be more sensitive to this overall impact. For example, the American Society of Clinical Oncology (ASCO) urged the USP organization to reevaluate the proposed USP <800> changes.6 ASCO noted the potential negative impact on access to cancer care because resources needed to implement these changes are not available in many oncology practice settings and represent prohibitive financial and administrative burdens on oncology practices and clinics.
REFERENCES
1. U.S. Pharmacopeial Convention. USP milestones—a timeline. www.usp.org/about-usp/our-history/usp-milestones-timeline. Accessed May 6, 2015.
2. U.S. Pharmacopeial Convention. Mission of the USP. www.usp.org/about-usp?destination=node %2F38. Accessed July 7, 2015.
3. USP Compounding Expert Committee. Proposed <800> Hazardous Drugs—Handling in Healthcare Settings. May-June 2013. www.usp.org/sites/default/files/usp_pdf/EN/m7808_pre-post.pdf. Accessed July 8, 2015.
4. Connor TH, MacKenzie BA, DeBord DG, et al. NIOSH List of Antineoplastic and Other Hazardous Drugs in Healthcare Settings, 2014. Cincinnati, OH: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health. DHHS (NIOSH) Publication No. 2014-138 (Supersedes 2012-150). September 2014. www.cdc.gov/niosh/docs/2014-138/pdfs/2014-138_v3.pdf. Accessed May 6, 2015.
5. ASTM International Standards (formerly known as the American Society for Testing and Materials). www.astm.org. Accessed May 6, 2015.
6. American Society of Clinical Oncology. ASCO concerned about proposed changes to USP requirements on handling hazardous drugs. August 1, 2014. www.asco.org/advocacy/asco-concerned-about-proposed-changes-usp-requirements-handling-hazardous-drugs. Accessed July 8, 2015.
To comment on this article, contact rdavidson@uspharmacist.com.



